Exemple
Le tracé d'arbre à partir de l'alignement proposé en téléchargement donne le résultat suivant :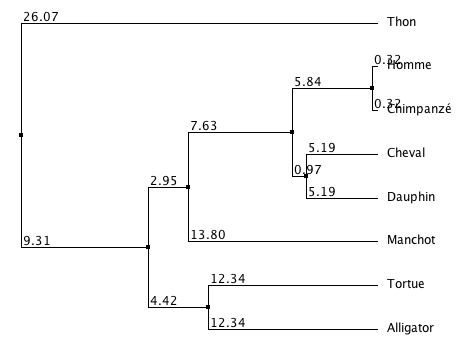 Remarque : les valeurs affichées sur les banches de l'arbre correspondent à une distance calculée en pourcentage de différence.
Remarque : les valeurs affichées sur les banches de l'arbre correspondent à une distance calculée en pourcentage de différence.
Pour supprimer ces valeurs de l'arbre, utiliser la commande View > Show distances
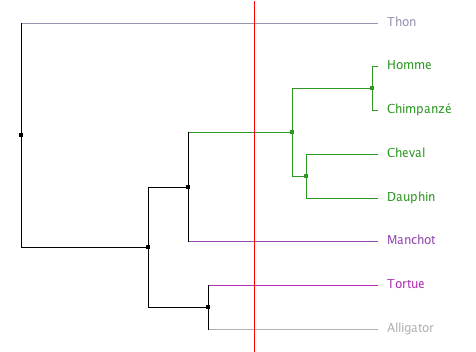 Un clic sur l'alignement a permit de tracer la ligne rouge verticale au niveau du pointeur de la souris
Un clic sur l'alignement a permit de tracer la ligne rouge verticale au niveau du pointeur de la souris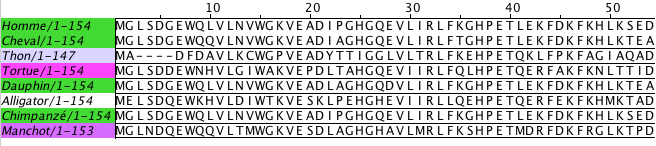 Sur l'alignement correspondant, les séquences ont été colorées de la même façon (d'une même couleur verte apparaissent les mammifères).
Sur l'alignement correspondant, les séquences ont été colorées de la même façon (d'une même couleur verte apparaissent les mammifères).





